
Overview
Mucopolysaccharidoses
MPS are a group of rare, inherited lysosomal storage disorders caused by a deficiency of a lysosomal enzyme responsible for breaking down glycoaminoglycans, or GAGs. The incidence of all MPS disorders is approximately 1:50,000 to 1:25,000. The incidence of MPS I is approximately 1:100,000.
Each of the MPS disorders are listed below. The table outlines the associated genes and enzyme deficiencies in each of the MPS disorders.
To read more on disease presentation, please visit our Publication page here.
The GAGs that accumulate in each of the MPS disorders vary. The table below highlights which GAGs are elevated in each of the MPS disorders.
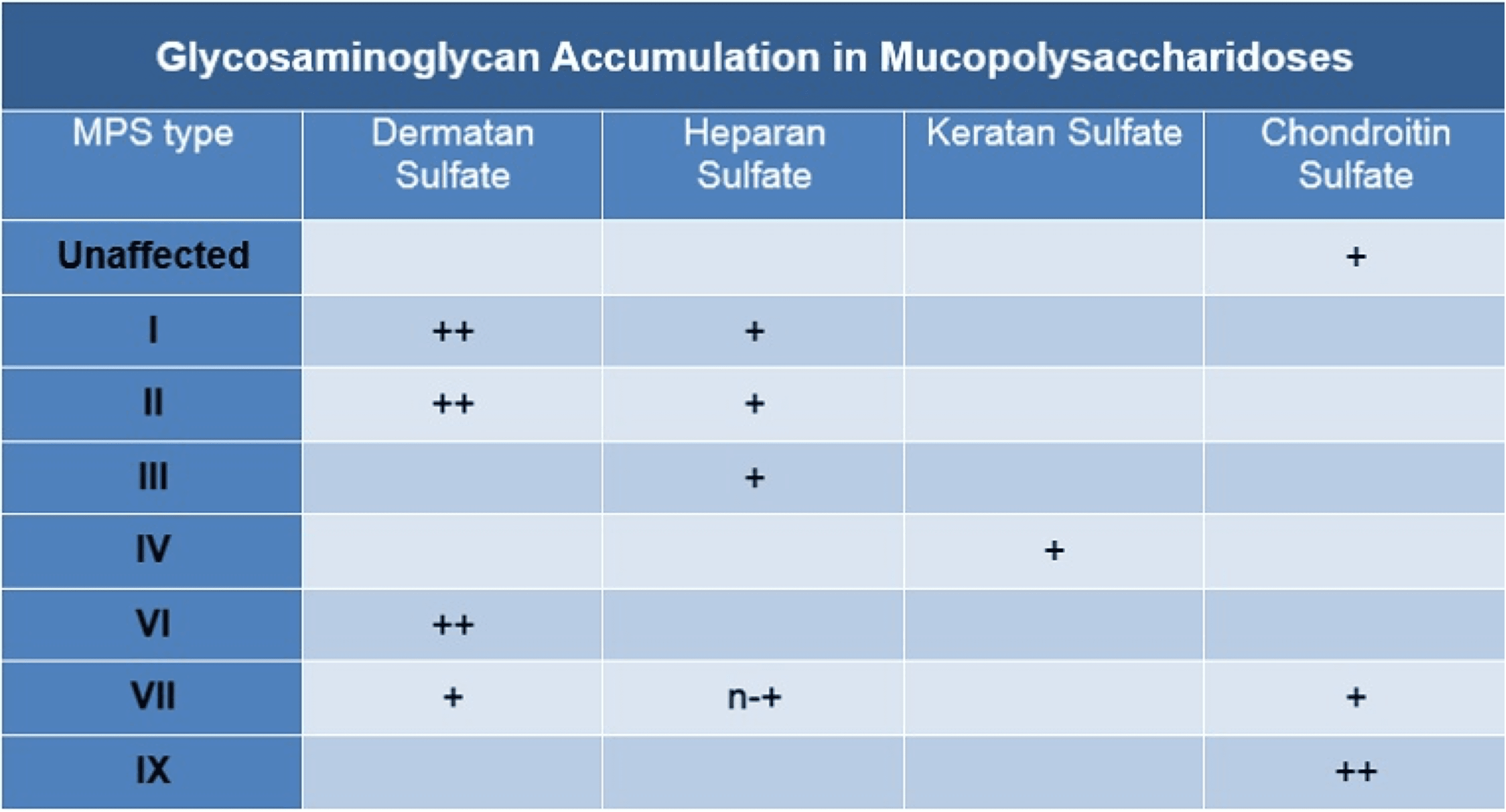
Adapted with permission from Zschocke/Hoffmann. Vademecum Metabolicum – Manual of Metabolic Paediatrics; 2nd Edition.
Mucopolysaccharidosis I (MPS I)
MPS I is 1 of the 7 mucopolysaccharidoses. It is an inherited, multi-system, progressive disorder caused by a deficiency of the lysosomal enzyme α-L-iduronidase, which is encoded by the IDUA gene. Pathogenic mutations to the IDUA gene lead to a defective enzyme with less than normal α-L-iduronidase activity. The decreased activity of this enzyme causes GAGs, particularly dermatan and heparan sulfate, to accumulate in the lysosome. Over time, the buildup of dermatan and heparan sulfate leads to excess cellular storage and cellular dysfunction, which ultimately leads to cell death and organ-specific clinical manifestations. The tissues and organs that are affected vary.

Systemic Effects of GAG Accumulation
The clinical severity of MPS I varies among patients. MPS I patients are commonly categorized into one of three clinical groups depending on their cognitive functioning. Patients with pronounced mental delays with loss of acquired skills are generally classified as Hurler or severe patients. Those with no to mild learning disabilities are commonly classified as Hurler-Scheie, and those with no cognitive impairments are classified as Scheie patients. Overall, all phenotypes across the MPS I spectrum are progressive and debilitating.
Multiple organ systems are affected in MPS I. The clinical presentation and organ involvement varies from patient to patient. Not
all patients will experience the same symptoms. The figure below outlines some of the clinical presentations that can present in
MPS I patients.

For a more in-depth look at epidemiology, inheritance, and pathophysiology, please look at our MPS 101 slides.
Or listen to a Medical Science Liaison discussing MPS I epidemiology, inheritance, manifestations, and diagnosis.
References:
Arn P, et al. J Pediatr. 2009;154(6)859–864.e3.; Clarke LA. NCBI Bookshelf, a service of the National Library of Medicine, National Institutes of Health (NIH). Available at: https://www.ncbi.nlm.nih.gov/books/NBK1162. Accessed July 20, 2018.
Muenzer J, et al. Pediatrics. 2009;123(1):19–29.; Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic & Molecular Bases of Inherited Disease. New York, NY: McGraw-Hill; 2001:3421–3452.; Vijay S, Wraith JE. Acta Paediatr. 2005;94(7):872–877.; National MPS Society. A guide to understanding MPS I. http://mpssociety.org/wp-content/uploads/2011/07/booklet_MPS_I_v8.pdf. 2008.
Next in MPS I